
8. Exemplo: ficheiros UniProt txt#
8.1. Introdução#
Este e o próximo capítulo são dedicados a aplicar as ferramentas mostradas na primeira parte a pequenos projetos já com alguma elaboração.
Os dois exemplos consistem em extraír informação relevante de ficheiros disponibilizados por portais e ferramentas de Bioinformática.
O primeiro exemplo consiste em extraír a informação sobre modificações pós-traducionais (em inglês: Post Translational Modifications, ou PTM) de um ficheiro contendo a informação da UniProt sobre todas as proteínas de um determinado organismo, um dos proteomas de referência existentes neste portal.
O que são PTMs? São modificações que ocorrem nas proteínas após a sua síntese. Uma das mais conhecidas é a fosforição de grupos laterais contendo -OH, que são muitas vezes são relacionadas com a regulação da função de uma proteína. Mas existem muitas outras modificações possíveis.
O objetivo é extraír a informação sobre PTMs e fazer uma contagem (e visualizar num gráfico) de modo a ter uma ideia da sua abundância relativa nas notações da UniProt.
O proteoma usado vai ser o do organismo Saccharomyces cerevisiae (a levedura de padeiro, um dos organismos modelo em biociências moleculares)
Este exemplo irá ilustrar muitas dos conceitos introduzidos nos capítulos anteriores, especialmente sobre a transformação de strings
Mas antes é necessário obter e preparar a informação total sobre as proteínas da levedura da UniProt.
8.2. Obtenção do ficheiro UniProt txt#
A UniProt disponibiliza vários tipos de ficheiros após a realização de uma busca.
Um dos mais completos são os ficheiros do tipo UniProt txt que contêm praticamente toda a informação que podemos visualizar quando consultamos as páginas web dedicadas a uma determinada proteína, mas num formato de texto e podendo juntar a informação de várias proteínas no mesmo ficheiro.
A UniProt tem documentação com indicações detalhadas sobre o formato dos seus ficheiros, em particular o UniProt txt:
Instruções para obter o ficheiro de trabalho:
Na UniProt procurar pelo “proteoma de referência” da levedura S. cerevisiae (http://www.uniprot.org/proteomes/UP000002311)
Passar para resultados UniProtKB: em “Map To” escolher UniPortKB
Escolher Download -> Text , compressed (NOTA: o ficheiro é grande, o download pode demorar um pouco)
Se o download tiver sido em modo “compressed”, extraír o (único) ficheiro do zip.
Alterar o nome do ficheiro para
uniprot_scerevisiae.txtCriar uma pasta de trabalho, onde irá ser desenvolvido o programa e mover o ficheiro
uniprot_scerevisiae.txtpara essa pasta.
8.3. Programa completo#
Comecemos por mostrar o programa completo e o respetivo output:
# Counts of the several types of PTMs in proteins of S. cerevisiae
from matplotlib import pyplot as plt
import seaborn as sns
data_filename = 'uniprot_scerevisiae.txt'
def read_Uniprot_text(filename):
"""Reads a UniProt text file and splits into a list of protein records."""
with open(filename) as uniprot_file:
whole_file = uniprot_file.read()
records = whole_file.split('//\n')
# remove empty records
records = [p for p in records if len(p) != 0]
# since we know that it is the last one only...
# records.pop(-1)
return records
prots = read_Uniprot_text(data_filename)
def extract_info(record):
"""Reads a UniProt text record and returns a dict with extrated information.
The returned dict has the following fields:
'ac': the UniProt Access Id,
'n': the sequence length,
'PTMs': a dictionary that associates the location of PTMs (int, as keys)
with the name of the PTM.
"""
IDline, ACline, *otherlines = record.splitlines()
# Extract UniProt AC and number of amino acids
# Example (first two lines):
# ID AB140_YEAST Reviewed; 628 AA.
# AC Q08641; D6W2U2; Q08644;
ac = ACline.split(';',1)[0].split()[1]
n = int(IDline.split()[3])
# Extract FT lines and process PTM information
#
# Example of phosphorylation lines
# FT MOD_RES 342
# FT /note="Phosphoserine"
# FT /evidence="ECO:0000244|PubMed:18407956,
# FT ECO:0000244|PubMed:19779198"
PTMs = {}
for i, line in enumerate(otherlines):
if line.startswith('FT MOD_RES'):
FTcode, MOD_RES, loc, *rest = line.split()
nextline = otherlines[i+1]
PTMtype = nextline.split('/note=')[1]
PTMtype = PTMtype.strip('"')
PTMtype = PTMtype.split(';')[0]
PTMs[loc] = PTMtype
# Return dictionary of extracted information
return {'ac': ac, 'n': n, 'PTMs': PTMs}
all_prots = [extract_info(p) for p in prots]
PTM_counts = {}
for p in all_prots:
PTMs = p['PTMs']
# just skip if no PTMs
if len(PTMs) == 0:
continue
for ptmtype in PTMs.values():
if ptmtype in PTM_counts:
PTM_counts[ptmtype] = PTM_counts[ptmtype] + 1
else:
PTM_counts[ptmtype] = 1
# sort function returning the second element of a pair of values
def second(pair):
return pair[1]
# sort items, using second element (the counts) as key, decreasing order
sorted_items = sorted(PTM_counts.items(), key=second, reverse=True)
# the result of sorted is not actually a list,
# but we can transform it into a list
ordered_PTM_counts = list(sorted_items)
print(f'The number of protein records in "{data_filename}" is {len(prots)}')
# let's look at the results
for PTMtype, count in ordered_PTM_counts:
print(PTMtype, count)
# make a barplot, only for PTMs with count > 10
sns.set(style="darkgrid")
f, ax = plt.subplots(figsize=(6,9))
types = [t for t,c in ordered_PTM_counts if c > 10]
counts = [c for t,c in ordered_PTM_counts if c > 10]
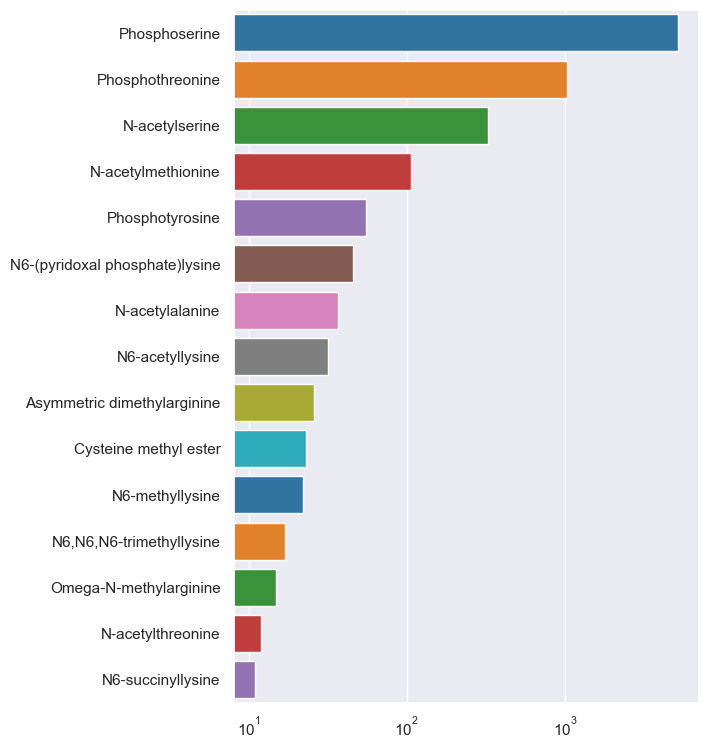
bp = sns.barplot(y=types, x=counts, orient='h', log=True, palette='tab10')
plt.show()
The number of protein records in "uniprot_scerevisiae.txt" is 6049
Phosphoserine 5170
Phosphothreonine 1027
N-acetylserine 325
N-acetylmethionine 106
Phosphotyrosine 55
N6-(pyridoxal phosphate)lysine 46
N-acetylalanine 37
N6-acetyllysine 32
Asymmetric dimethylarginine 26
Cysteine methyl ester 23
N6-methyllysine 22
N6,N6,N6-trimethyllysine 17
Omega-N-methylarginine 15
N-acetylthreonine 12
N6-succinyllysine 11
N6,N6-dimethyllysine 6
N6-butyryllysine 5
N6-biotinyllysine 5
N6-carboxylysine 4
Phosphohistidine 4
N5-methylglutamine 4
N,N-dimethylproline 4
N6-lipoyllysine 4
O-(pantetheine 4'-phosphoryl)serine 3
4-aspartylphosphate 3
Lysine derivative 3
Pyruvic acid (Ser) 3
S-glutathionyl cysteine 3
Hypusine 2
Tele-8alpha-FAD histidine 2
N6-malonyllysine 2
N5-methylarginine 2
N6-propionyllysine 2
Leucine methyl ester 2
3,4-dihydroxyproline 2
N-acetylvaline 2
N-formylmethionine 1
Pros-8alpha-FAD histidine 1
2,3-didehydroalanine (Cys) 1
N6-glutaryllysine 1
Diphthamide 1
Pros-methylhistidine 1
Cysteine sulfinic acid (-SO2H) 1
S-(dipyrrolylmethanemethyl)cysteine 1
N,N,N-trimethylglycine 1
Lysine methyl ester 1
Dimethylated arginine 1
Thiazolidine linkage to a ring-opened DNA abasic 1
1-thioglycine 1
S-methylcysteine 1
De uma forma sumária, o programa realiza as seguintes tarefas:
leitura do ficheiro
uniprot_scerevisiae.txtpara uma string e separação da informação por proteína, criando a listaprotsExtração da informação, criando uma lista de dicionários,
all_prots, em que cada dicionário tem o número de acesso, comprimento da proteína e tabela de PTMsContagem dos diferentes tipos de PTM, ordenando as contagens por ordem decrescente
Apresentação dos resultados, incluíndo um gráfico de barras
As duas primeiras tarefas são implementadas em funções.
Para o gráfico, são usados dois módulos de criação de gráficos da linguagem Python: matplotlib e seaborn. Eles são importados no início do programa. É prática comum que os imports sejam feitos no início do programa.
Vejamos agora as várias partes:
8.4. Leitura do ficheiro e separação da informação por proteína#
Para o programa funcionar, o ficheiro uniprot_scerevisiae.txt deve estar na mesma pasta que o programa.
Podemos começar por ler todo o seu conteúdo para uma string:
data_filename = 'uniprot_scerevisiae.txt'
with open(data_filename) as uniprot_file:
whole_file = uniprot_file.read()
O passo seguinte será, na string whole_file, que contem toda a informação sobre todas as proteínas de levedura, separarmos a informação por proteína, pondo o resultado numa lista de strings, blocos de texto em que cada bloco diz respeito a uma proteína diferente.
Para isso precisamos de saber o que “divide”, o que separa no texto as proteínas umas das outras. Isto na perspetiva de usarmos a função .split() para separarmos a informação a partir de um separador, criando uma lista.
Qual é o separador?
Podemos abrir o ficheiro num editor de texto e reparamos nos pormenores da sua estrutura.
Depressa nos apercebemos que existe uma marca para separar a informação de diferentes proteínas:
...
...
FT CARBOHYD 103 103 N-linked (GlcNAc...) asparagine.
FT {ECO:0000255}.
SQ SEQUENCE 411 AA; 48455 MW; 91676D56AC053F3C CRC64;
MTSATDKSID RLVVNAKTRR RNSSVGKIDL GDTVPGFAAM PESAASKNEA KKRMKALTGD
SKKDSDLLWK VWFSYREMNY RHSWLTPFFI LVCVYSAYFL SGNRTESNPL HMFVAISYQV
DGTDSYAKGI KDLSFVFFYM IFFTFLREFL MDVVIRPFTV YLNVTSEHRQ KRMLEQMYAI
FYCGVSGPFG LYIMYHSDLW LFKTKPMYRT YPVITNPFLF KIFYLGQAAF WAQQACVLVL
QLEKPRKDYK ELVFHHIVTL LLIWSSYVFH FTKMGLAIYI TMDVSDFFLS LSKTLNYLNS
VFTPFVFGLF VFFWIYLRHV VNIRILWSVL TEFRHEGNYV LNFATQQYKC WISLPIVFVL
IAALQLVNLY WLFLILRILY RLIWQGIQKD ERSDSDSDES AENEESKEKC E
//
ID MUD2_YEAST Reviewed; 527 AA.
AC P36084; D6VXL2;
DT 01-JUN-1994, integrated into UniProtKB/Swiss-Prot.
DT 16-AUG-2004, sequence version 3.
DT 13-FEB-2019, entry version 153.
DE RecName: Full=Splicing factor MUD2;
...
...
Informação
No formato Uniprot txt o separador que aparece entre a informação sobre proteínas distintas é // numa
linha.
Não deve existir mais nada nessa linha, o que significa que o separador é, na realidade, // seguido de mudança de linha, isto é, //\n
Portanto, podemos dividir todo o ficheiro em blocos,
indicando //\n na função .split():
data_filename = 'uniprot_scerevisiae.txt'
with open(data_filename) as uniprot_file:
whole_file = uniprot_file.read()
records = whole_file.split('//\n')
Podemos ver se resultou.
Como records, o nome dado ao resultado de split('//\n') é uma lista, podemos ver o elemento 0, por exemplo:
data_filename = 'uniprot_scerevisiae.txt'
with open(data_filename) as uniprot_file:
whole_file = uniprot_file.read()
records = whole_file.split('//\n')
print(records[0])
ID AGP2_YEAST Reviewed; 596 AA.
AC P38090; D6VQC9;
DT 01-OCT-1994, integrated into UniProtKB/Swiss-Prot.
DT 01-OCT-1994, sequence version 1.
DT 22-APR-2020, entry version 155.
DE RecName: Full=General amino acid permease AGP2;
GN Name=AGP2; OrderedLocusNames=YBR132C; ORFNames=YBR1007;
OS Saccharomyces cerevisiae (strain ATCC 204508 / S288c) (Baker's yeast).
OC Eukaryota; Fungi; Dikarya; Ascomycota; Saccharomycotina; Saccharomycetes;
OC Saccharomycetales; Saccharomycetaceae; Saccharomyces.
OX NCBI_TaxID=559292;
RN [1]
RP NUCLEOTIDE SEQUENCE [GENOMIC DNA].
RC STRAIN=ATCC 204508 / S288c;
RX PubMed=8091856; DOI=10.1002/yea.320100002;
RA Becam A.-M., Cullin C., Grzybowska E., Lacroute F., Nasr F.,
RA Ozier-Kalogeropoulos O., Palucha A., Slonimski P.P., Zagulski M.,
RA Herbert C.J.;
RT "The sequence of 29.7 kb from the right arm of chromosome II reveals 13
RT complete open reading frames, of which ten correspond to new genes.";
RL Yeast 10:S1-S11(1994).
RN [2]
RP NUCLEOTIDE SEQUENCE [LARGE SCALE GENOMIC DNA].
RC STRAIN=ATCC 204508 / S288c;
RX PubMed=7813418; DOI=10.1002/j.1460-2075.1994.tb06923.x;
RA Feldmann H., Aigle M., Aljinovic G., Andre B., Baclet M.C., Barthe C.,
RA Baur A., Becam A.-M., Biteau N., Boles E., Brandt T., Brendel M.,
RA Brueckner M., Bussereau F., Christiansen C., Contreras R., Crouzet M.,
RA Cziepluch C., Demolis N., Delaveau T., Doignon F., Domdey H.,
RA Duesterhus S., Dubois E., Dujon B., El Bakkoury M., Entian K.-D.,
RA Feuermann M., Fiers W., Fobo G.M., Fritz C., Gassenhuber J., Glansdorff N.,
RA Goffeau A., Grivell L.A., de Haan M., Hein C., Herbert C.J.,
RA Hollenberg C.P., Holmstroem K., Jacq C., Jacquet M., Jauniaux J.-C.,
RA Jonniaux J.-L., Kallesoee T., Kiesau P., Kirchrath L., Koetter P.,
RA Korol S., Liebl S., Logghe M., Lohan A.J.E., Louis E.J., Li Z.Y.,
RA Maat M.J., Mallet L., Mannhaupt G., Messenguy F., Miosga T., Molemans F.,
RA Mueller S., Nasr F., Obermaier B., Perea J., Pierard A., Piravandi E.,
RA Pohl F.M., Pohl T.M., Potier S., Proft M., Purnelle B., Ramezani Rad M.,
RA Rieger M., Rose M., Schaaff-Gerstenschlaeger I., Scherens B.,
RA Schwarzlose C., Skala J., Slonimski P.P., Smits P.H.M., Souciet J.-L.,
RA Steensma H.Y., Stucka R., Urrestarazu L.A., van der Aart Q.J.M.,
RA Van Dyck L., Vassarotti A., Vetter I., Vierendeels F., Vissers S.,
RA Wagner G., de Wergifosse P., Wolfe K.H., Zagulski M., Zimmermann F.K.,
RA Mewes H.-W., Kleine K.;
RT "Complete DNA sequence of yeast chromosome II.";
RL EMBO J. 13:5795-5809(1994).
RN [3]
RP GENOME REANNOTATION.
RC STRAIN=ATCC 204508 / S288c;
RX PubMed=24374639; DOI=10.1534/g3.113.008995;
RA Engel S.R., Dietrich F.S., Fisk D.G., Binkley G., Balakrishnan R.,
RA Costanzo M.C., Dwight S.S., Hitz B.C., Karra K., Nash R.S., Weng S.,
RA Wong E.D., Lloyd P., Skrzypek M.S., Miyasato S.R., Simison M., Cherry J.M.;
RT "The reference genome sequence of Saccharomyces cerevisiae: Then and now.";
RL G3 (Bethesda) 4:389-398(2014).
RN [4]
RP DISCUSSION OF SEQUENCE.
RX PubMed=7954890; DOI=10.1007/bf00326297;
RA Nasr F., Becam A.-M., Grzybowska E., Zagulski M., Slonimski P.P.,
RA Herbert C.J.;
RT "An analysis of the sequence of part of the right arm of chromosome II of
RT S. cerevisiae reveals new genes encoding an amino-acid permease and a
RT carboxypeptidase.";
RL Curr. Genet. 26:1-7(1994).
RN [5]
RP CHARACTERIZATION.
RA Garrett J.M., Schreve J.L.;
RL Unpublished observations (JUL-1997).
RN [6]
RP FUNCTION IN CARNITINE TRANSPORT.
RX PubMed=10545096; DOI=10.1093/emboj/18.21.5843;
RA van Roermund C.W., Hettema E.H., van den Berg M., Tabak H.F., Wanders R.J.;
RT "Molecular characterization of carnitine-dependent transport of acetyl-CoA
RT from peroxisomes to mitochondria in Saccharomyces cerevisiae and
RT identification of a plasma membrane carnitine transporter, Agp2p.";
RL EMBO J. 18:5843-5852(1999).
RN [7]
RP FUNCTION IN CARNITINE TRANSPORT.
RX PubMed=12132580;
RA Lee J., Lee B., Shin D., Kwak S.S., Bahk J.D., Lim C.O., Yun D.J.;
RT "Carnitine uptake by AGP2 in yeast Saccharomyces cerevisiae is dependent on
RT Hog1 MAP kinase pathway.";
RL Mol. Cells 13:407-412(2002).
RN [8]
RP TOPOLOGY [LARGE SCALE ANALYSIS].
RC STRAIN=ATCC 208353 / W303-1A;
RX PubMed=16847258; DOI=10.1073/pnas.0604075103;
RA Kim H., Melen K., Oesterberg M., von Heijne G.;
RT "A global topology map of the Saccharomyces cerevisiae membrane proteome.";
RL Proc. Natl. Acad. Sci. U.S.A. 103:11142-11147(2006).
RN [9]
RP IDENTIFICATION BY MASS SPECTROMETRY [LARGE SCALE ANALYSIS].
RX PubMed=19779198; DOI=10.1126/science.1172867;
RA Holt L.J., Tuch B.B., Villen J., Johnson A.D., Gygi S.P., Morgan D.O.;
RT "Global analysis of Cdk1 substrate phosphorylation sites provides insights
RT into evolution.";
RL Science 325:1682-1686(2009).
CC -!- FUNCTION: General amino acid permease with broad substrate specificity.
CC Can also transport carnitine. {ECO:0000269|PubMed:10545096,
CC ECO:0000269|PubMed:12132580}.
CC -!- INTERACTION:
CC P38090; P43603: LSB3; NbExp=2; IntAct=EBI-2362, EBI-22980;
CC P38090; P32793: YSC84; NbExp=2; IntAct=EBI-2362, EBI-24460;
CC -!- SUBCELLULAR LOCATION: Membrane; Multi-pass membrane protein.
CC -!- SIMILARITY: Belongs to the amino acid-polyamine-organocation (APC)
CC superfamily. YAT (TC 2.A.3.10) family. {ECO:0000305}.
CC ---------------------------------------------------------------------------
CC Copyrighted by the UniProt Consortium, see https://www.uniprot.org/terms
CC Distributed under the Creative Commons Attribution (CC BY 4.0) License
CC ---------------------------------------------------------------------------
DR EMBL; X75891; CAA53491.1; -; Genomic_DNA.
DR EMBL; Z36001; CAA85089.1; -; Genomic_DNA.
DR EMBL; BK006936; DAA07249.1; -; Genomic_DNA.
DR PIR; S46001; S46001.
DR RefSeq; NP_009690.1; NM_001178480.1.
DR BioGrid; 32833; 117.
DR DIP; DIP-8141N; -.
DR IntAct; P38090; 8.
DR MINT; P38090; -.
DR STRING; 4932.YBR132C; -.
DR TCDB; 2.A.3.10.19; the amino acid-polyamine-organocation (apc) family.
DR iPTMnet; P38090; -.
DR MaxQB; P38090; -.
DR PaxDb; P38090; -.
DR PRIDE; P38090; -.
DR EnsemblFungi; YBR132C_mRNA; YBR132C; YBR132C.
DR GeneID; 852429; -.
DR KEGG; sce:YBR132C; -.
DR EuPathDB; FungiDB:YBR132C; -.
DR SGD; S000000336; AGP2.
DR HOGENOM; CLU_007946_12_1_1; -.
DR InParanoid; P38090; -.
DR KO; K16261; -.
DR OMA; NFWFLEC; -.
DR BioCyc; YEAST:G3O-29087-MONOMER; -.
DR SABIO-RK; P38090; -.
DR PRO; PR:P38090; -.
DR Proteomes; UP000002311; Chromosome II.
DR RNAct; P38090; protein.
DR GO; GO:0005783; C:endoplasmic reticulum; HDA:SGD.
DR GO; GO:0005789; C:endoplasmic reticulum membrane; IDA:SGD.
DR GO; GO:0000329; C:fungal-type vacuole membrane; IDA:SGD.
DR GO; GO:0016021; C:integral component of membrane; IBA:GO_Central.
DR GO; GO:0005887; C:integral component of plasma membrane; IDA:SGD.
DR GO; GO:0015171; F:amino acid transmembrane transporter activity; IBA:GO_Central.
DR GO; GO:0003333; P:amino acid transmembrane transport; IBA:GO_Central.
DR GO; GO:1902274; P:positive regulation of (R)-carnitine transmembrane transport; IMP:SGD.
DR GO; GO:1902269; P:positive regulation of polyamine transmembrane transport; IMP:SGD.
DR InterPro; IPR004841; AA-permease/SLC12A_dom.
DR InterPro; IPR002293; AA/rel_permease1.
DR InterPro; IPR004840; Amoino_acid_permease_CS.
DR Pfam; PF00324; AA_permease; 1.
DR PIRSF; PIRSF006060; AA_transporter; 1.
DR PROSITE; PS00218; AMINO_ACID_PERMEASE_1; 1.
PE 1: Evidence at protein level;
KW Amino-acid transport; Membrane; Reference proteome; Transmembrane;
KW Transmembrane helix; Transport.
FT CHAIN 1..596
FT /note="General amino acid permease AGP2"
FT /id="PRO_0000054143"
FT TOPO_DOM 1..94
FT /note="Cytoplasmic"
FT /evidence="ECO:0000255"
FT TRANSMEM 95..115
FT /note="Helical"
FT /evidence="ECO:0000255"
FT TOPO_DOM 116..117
FT /note="Extracellular"
FT /evidence="ECO:0000255"
FT TRANSMEM 118..138
FT /note="Helical"
FT /evidence="ECO:0000255"
FT TOPO_DOM 139..165
FT /note="Cytoplasmic"
FT /evidence="ECO:0000255"
FT TRANSMEM 166..186
FT /note="Helical"
FT /evidence="ECO:0000255"
FT TOPO_DOM 187..199
FT /note="Extracellular"
FT /evidence="ECO:0000255"
FT TRANSMEM 200..220
FT /note="Helical"
FT /evidence="ECO:0000255"
FT TOPO_DOM 221..227
FT /note="Cytoplasmic"
FT /evidence="ECO:0000255"
FT TRANSMEM 228..248
FT /note="Helical"
FT /evidence="ECO:0000255"
FT TOPO_DOM 249..288
FT /note="Extracellular"
FT /evidence="ECO:0000255"
FT TRANSMEM 289..309
FT /note="Helical"
FT /evidence="ECO:0000255"
FT TOPO_DOM 310..326
FT /note="Cytoplasmic"
FT /evidence="ECO:0000255"
FT TRANSMEM 327..347
FT /note="Helical"
FT /evidence="ECO:0000255"
FT TOPO_DOM 348..379
FT /note="Extracellular"
FT /evidence="ECO:0000255"
FT TRANSMEM 380..400
FT /note="Helical"
FT /evidence="ECO:0000255"
FT TOPO_DOM 401..427
FT /note="Cytoplasmic"
FT /evidence="ECO:0000255"
FT TRANSMEM 428..448
FT /note="Helical"
FT /evidence="ECO:0000255"
FT TOPO_DOM 449..459
FT /note="Extracellular"
FT /evidence="ECO:0000255"
FT TRANSMEM 460..480
FT /note="Helical"
FT /evidence="ECO:0000255"
FT TOPO_DOM 481..505
FT /note="Cytoplasmic"
FT /evidence="ECO:0000255"
FT TRANSMEM 506..526
FT /note="Helical"
FT /evidence="ECO:0000255"
FT TOPO_DOM 527..534
FT /note="Extracellular"
FT /evidence="ECO:0000255"
FT TRANSMEM 535..555
FT /note="Helical"
FT /evidence="ECO:0000255"
FT TOPO_DOM 556..596
FT /note="Cytoplasmic"
FT /evidence="ECO:0000255"
SQ SEQUENCE 596 AA; 67262 MW; 8658DCDA5A7D6B64 CRC64;
MTKERMTIDY ENDGDFEYDK NKYKTITTRI KSIEPSEGWL EPSGSVGHIN TIPEAGDVHV
DEHEDRGSSI DDDSRTYLLY FTETRRKLEN RHVQLIAISG VIGTALFVAI GKALYRGGPA
SLLLAFALWC VPILCITVST AEMVCFFPVS SPFLRLATKC VDDSLAVMAS WNFWFLECVQ
IPFEIVSVNT IIHYWRDDYS AGIPLAVQVV LYLLISICAV KYYGEMEFWL ASFKIILALG
LFTFTFITML GGNPEHDRYG FRNYGESPFK KYFPDGNDVG KSSGYFQGFL ACLIQASFTI
AGGEYISMLA GEVKRPRKVL PKAFKQVFVR LTFLFLGSCL CVGIVCSPND PDLTAAINEA
RPGAGSSPYV IAMNNLKIRI LPDIVNIALI TAAFSAGNAY TYCSSRTFYG MALDGYAPKI
FTRCNRHGVP IYSVAISLVW ALVSLLQLNS NSAVVLNWLI NLITASQLIN FVVLCIVYLF
FRRAYHVQQD SLPKLPFRSW GQPYTAIIGL VSCSAMILIQ GYTVFFPKLW NTQDFLFSYL
MVFINIGIYV GYKFIWKRGK DHFKNPHEID FSKELTEIEN HEIESSFEKF QYYSKA
Parece ter resultado.
Ainda para confirmar podemos ver o último elemento da lista (posição -1):
data_filename = 'uniprot_scerevisiae.txt'
with open(data_filename) as uniprot_file:
whole_file = uniprot_file.read()
records = whole_file.split('//\n')
print(records[-1])
Desta vez nada apareceu como resultado da função print().
Porquê?
É apenas um pequeno problema que resulta do facto do ficheiro acabar com uma linha contendo //. (Por favor confirmar)
Isto faz com que o resultado da função split() tenha, no final da lista, uma string vazia.
Esta string vazia deve ser retirada do fim da lista.
Podemos usar uma lista em compreensão para a retirar:
records = [p for p in records if len(p) != 0]
isto é, mantemos todos os records, desde que o seu comprimento seja maior do que 0, isto é, não seja uma lista vazia.
Outra possibilidade é usar a função pop() de listas que
retira o elemento que está numa posição:
records.pop(-1)
Esta “tarefa” pode ser posta numa função, uma vez que é uma parte lógica do programa (ler o ficheiro e dividir em blocos respeitantes a diferentes proteínas).
Podemos definir a função aplicar a função logo de seguida.
Como curiosidade, podemos saber qual a dimensão deste proteoma anotado da levedura:
def read_Uniprot_text(filename):
"""Reads a UniProt text file and splits into a list of protein records."""
with open(filename) as uniprot_file:
whole_file = uniprot_file.read()
records = whole_file.split('//\n')
# remove empty records
records = [p for p in records if len(p) != 0]
# since we know that it is the last one only...
# records.pop(-1)
return records
prots = read_Uniprot_text(data_filename)
print(f'The number of protein records in "{data_filename}" is {len(prots)}')
The number of protein records in "uniprot_scerevisiae.txt" is 6049
8.5. Extração da informação#
O interesse agora é extrair a informação pertinente de cada proteína.
Estamos interessados em informações sobre as PTMs. Já vamos ver onde essa informação se encontra e em que formato está. Mas para treinar a extração da informação, podemos começar por tentar obter, para cada proteína, o seu número de acesso Uniprot, umm identificador único para cada proteína e o número de aminoácidos da proteína.
A ideia é conseguir criar um dicionário com estes dois “pedaços” de informação.
Vejamos melhor a primeira proteína da lista, prots[0]:
# NOTA: continuação do programa anterior!
print(prots[0])
ID AGP2_YEAST Reviewed; 596 AA.
AC P38090; D6VQC9;
DT 01-OCT-1994, integrated into UniProtKB/Swiss-Prot.
DT 01-OCT-1994, sequence version 1.
DT 22-APR-2020, entry version 155.
DE RecName: Full=General amino acid permease AGP2;
GN Name=AGP2; OrderedLocusNames=YBR132C; ORFNames=YBR1007;
OS Saccharomyces cerevisiae (strain ATCC 204508 / S288c) (Baker's yeast).
OC Eukaryota; Fungi; Dikarya; Ascomycota; Saccharomycotina; Saccharomycetes;
OC Saccharomycetales; Saccharomycetaceae; Saccharomyces.
OX NCBI_TaxID=559292;
RN [1]
RP NUCLEOTIDE SEQUENCE [GENOMIC DNA].
RC STRAIN=ATCC 204508 / S288c;
RX PubMed=8091856; DOI=10.1002/yea.320100002;
RA Becam A.-M., Cullin C., Grzybowska E., Lacroute F., Nasr F.,
RA Ozier-Kalogeropoulos O., Palucha A., Slonimski P.P., Zagulski M.,
RA Herbert C.J.;
RT "The sequence of 29.7 kb from the right arm of chromosome II reveals 13
RT complete open reading frames, of which ten correspond to new genes.";
RL Yeast 10:S1-S11(1994).
RN [2]
RP NUCLEOTIDE SEQUENCE [LARGE SCALE GENOMIC DNA].
RC STRAIN=ATCC 204508 / S288c;
RX PubMed=7813418; DOI=10.1002/j.1460-2075.1994.tb06923.x;
RA Feldmann H., Aigle M., Aljinovic G., Andre B., Baclet M.C., Barthe C.,
RA Baur A., Becam A.-M., Biteau N., Boles E., Brandt T., Brendel M.,
RA Brueckner M., Bussereau F., Christiansen C., Contreras R., Crouzet M.,
RA Cziepluch C., Demolis N., Delaveau T., Doignon F., Domdey H.,
RA Duesterhus S., Dubois E., Dujon B., El Bakkoury M., Entian K.-D.,
RA Feuermann M., Fiers W., Fobo G.M., Fritz C., Gassenhuber J., Glansdorff N.,
RA Goffeau A., Grivell L.A., de Haan M., Hein C., Herbert C.J.,
RA Hollenberg C.P., Holmstroem K., Jacq C., Jacquet M., Jauniaux J.-C.,
RA Jonniaux J.-L., Kallesoee T., Kiesau P., Kirchrath L., Koetter P.,
RA Korol S., Liebl S., Logghe M., Lohan A.J.E., Louis E.J., Li Z.Y.,
RA Maat M.J., Mallet L., Mannhaupt G., Messenguy F., Miosga T., Molemans F.,
RA Mueller S., Nasr F., Obermaier B., Perea J., Pierard A., Piravandi E.,
RA Pohl F.M., Pohl T.M., Potier S., Proft M., Purnelle B., Ramezani Rad M.,
RA Rieger M., Rose M., Schaaff-Gerstenschlaeger I., Scherens B.,
RA Schwarzlose C., Skala J., Slonimski P.P., Smits P.H.M., Souciet J.-L.,
RA Steensma H.Y., Stucka R., Urrestarazu L.A., van der Aart Q.J.M.,
RA Van Dyck L., Vassarotti A., Vetter I., Vierendeels F., Vissers S.,
RA Wagner G., de Wergifosse P., Wolfe K.H., Zagulski M., Zimmermann F.K.,
RA Mewes H.-W., Kleine K.;
RT "Complete DNA sequence of yeast chromosome II.";
RL EMBO J. 13:5795-5809(1994).
RN [3]
RP GENOME REANNOTATION.
RC STRAIN=ATCC 204508 / S288c;
RX PubMed=24374639; DOI=10.1534/g3.113.008995;
RA Engel S.R., Dietrich F.S., Fisk D.G., Binkley G., Balakrishnan R.,
RA Costanzo M.C., Dwight S.S., Hitz B.C., Karra K., Nash R.S., Weng S.,
RA Wong E.D., Lloyd P., Skrzypek M.S., Miyasato S.R., Simison M., Cherry J.M.;
RT "The reference genome sequence of Saccharomyces cerevisiae: Then and now.";
RL G3 (Bethesda) 4:389-398(2014).
RN [4]
RP DISCUSSION OF SEQUENCE.
RX PubMed=7954890; DOI=10.1007/bf00326297;
RA Nasr F., Becam A.-M., Grzybowska E., Zagulski M., Slonimski P.P.,
RA Herbert C.J.;
RT "An analysis of the sequence of part of the right arm of chromosome II of
RT S. cerevisiae reveals new genes encoding an amino-acid permease and a
RT carboxypeptidase.";
RL Curr. Genet. 26:1-7(1994).
RN [5]
RP CHARACTERIZATION.
RA Garrett J.M., Schreve J.L.;
RL Unpublished observations (JUL-1997).
RN [6]
RP FUNCTION IN CARNITINE TRANSPORT.
RX PubMed=10545096; DOI=10.1093/emboj/18.21.5843;
RA van Roermund C.W., Hettema E.H., van den Berg M., Tabak H.F., Wanders R.J.;
RT "Molecular characterization of carnitine-dependent transport of acetyl-CoA
RT from peroxisomes to mitochondria in Saccharomyces cerevisiae and
RT identification of a plasma membrane carnitine transporter, Agp2p.";
RL EMBO J. 18:5843-5852(1999).
RN [7]
RP FUNCTION IN CARNITINE TRANSPORT.
RX PubMed=12132580;
RA Lee J., Lee B., Shin D., Kwak S.S., Bahk J.D., Lim C.O., Yun D.J.;
RT "Carnitine uptake by AGP2 in yeast Saccharomyces cerevisiae is dependent on
RT Hog1 MAP kinase pathway.";
RL Mol. Cells 13:407-412(2002).
RN [8]
RP TOPOLOGY [LARGE SCALE ANALYSIS].
RC STRAIN=ATCC 208353 / W303-1A;
RX PubMed=16847258; DOI=10.1073/pnas.0604075103;
RA Kim H., Melen K., Oesterberg M., von Heijne G.;
RT "A global topology map of the Saccharomyces cerevisiae membrane proteome.";
RL Proc. Natl. Acad. Sci. U.S.A. 103:11142-11147(2006).
RN [9]
RP IDENTIFICATION BY MASS SPECTROMETRY [LARGE SCALE ANALYSIS].
RX PubMed=19779198; DOI=10.1126/science.1172867;
RA Holt L.J., Tuch B.B., Villen J., Johnson A.D., Gygi S.P., Morgan D.O.;
RT "Global analysis of Cdk1 substrate phosphorylation sites provides insights
RT into evolution.";
RL Science 325:1682-1686(2009).
CC -!- FUNCTION: General amino acid permease with broad substrate specificity.
CC Can also transport carnitine. {ECO:0000269|PubMed:10545096,
CC ECO:0000269|PubMed:12132580}.
CC -!- INTERACTION:
CC P38090; P43603: LSB3; NbExp=2; IntAct=EBI-2362, EBI-22980;
CC P38090; P32793: YSC84; NbExp=2; IntAct=EBI-2362, EBI-24460;
CC -!- SUBCELLULAR LOCATION: Membrane; Multi-pass membrane protein.
CC -!- SIMILARITY: Belongs to the amino acid-polyamine-organocation (APC)
CC superfamily. YAT (TC 2.A.3.10) family. {ECO:0000305}.
CC ---------------------------------------------------------------------------
CC Copyrighted by the UniProt Consortium, see https://www.uniprot.org/terms
CC Distributed under the Creative Commons Attribution (CC BY 4.0) License
CC ---------------------------------------------------------------------------
DR EMBL; X75891; CAA53491.1; -; Genomic_DNA.
DR EMBL; Z36001; CAA85089.1; -; Genomic_DNA.
DR EMBL; BK006936; DAA07249.1; -; Genomic_DNA.
DR PIR; S46001; S46001.
DR RefSeq; NP_009690.1; NM_001178480.1.
DR BioGrid; 32833; 117.
DR DIP; DIP-8141N; -.
DR IntAct; P38090; 8.
DR MINT; P38090; -.
DR STRING; 4932.YBR132C; -.
DR TCDB; 2.A.3.10.19; the amino acid-polyamine-organocation (apc) family.
DR iPTMnet; P38090; -.
DR MaxQB; P38090; -.
DR PaxDb; P38090; -.
DR PRIDE; P38090; -.
DR EnsemblFungi; YBR132C_mRNA; YBR132C; YBR132C.
DR GeneID; 852429; -.
DR KEGG; sce:YBR132C; -.
DR EuPathDB; FungiDB:YBR132C; -.
DR SGD; S000000336; AGP2.
DR HOGENOM; CLU_007946_12_1_1; -.
DR InParanoid; P38090; -.
DR KO; K16261; -.
DR OMA; NFWFLEC; -.
DR BioCyc; YEAST:G3O-29087-MONOMER; -.
DR SABIO-RK; P38090; -.
DR PRO; PR:P38090; -.
DR Proteomes; UP000002311; Chromosome II.
DR RNAct; P38090; protein.
DR GO; GO:0005783; C:endoplasmic reticulum; HDA:SGD.
DR GO; GO:0005789; C:endoplasmic reticulum membrane; IDA:SGD.
DR GO; GO:0000329; C:fungal-type vacuole membrane; IDA:SGD.
DR GO; GO:0016021; C:integral component of membrane; IBA:GO_Central.
DR GO; GO:0005887; C:integral component of plasma membrane; IDA:SGD.
DR GO; GO:0015171; F:amino acid transmembrane transporter activity; IBA:GO_Central.
DR GO; GO:0003333; P:amino acid transmembrane transport; IBA:GO_Central.
DR GO; GO:1902274; P:positive regulation of (R)-carnitine transmembrane transport; IMP:SGD.
DR GO; GO:1902269; P:positive regulation of polyamine transmembrane transport; IMP:SGD.
DR InterPro; IPR004841; AA-permease/SLC12A_dom.
DR InterPro; IPR002293; AA/rel_permease1.
DR InterPro; IPR004840; Amoino_acid_permease_CS.
DR Pfam; PF00324; AA_permease; 1.
DR PIRSF; PIRSF006060; AA_transporter; 1.
DR PROSITE; PS00218; AMINO_ACID_PERMEASE_1; 1.
PE 1: Evidence at protein level;
KW Amino-acid transport; Membrane; Reference proteome; Transmembrane;
KW Transmembrane helix; Transport.
FT CHAIN 1..596
FT /note="General amino acid permease AGP2"
FT /id="PRO_0000054143"
FT TOPO_DOM 1..94
FT /note="Cytoplasmic"
FT /evidence="ECO:0000255"
FT TRANSMEM 95..115
FT /note="Helical"
FT /evidence="ECO:0000255"
FT TOPO_DOM 116..117
FT /note="Extracellular"
FT /evidence="ECO:0000255"
FT TRANSMEM 118..138
FT /note="Helical"
FT /evidence="ECO:0000255"
FT TOPO_DOM 139..165
FT /note="Cytoplasmic"
FT /evidence="ECO:0000255"
FT TRANSMEM 166..186
FT /note="Helical"
FT /evidence="ECO:0000255"
FT TOPO_DOM 187..199
FT /note="Extracellular"
FT /evidence="ECO:0000255"
FT TRANSMEM 200..220
FT /note="Helical"
FT /evidence="ECO:0000255"
FT TOPO_DOM 221..227
FT /note="Cytoplasmic"
FT /evidence="ECO:0000255"
FT TRANSMEM 228..248
FT /note="Helical"
FT /evidence="ECO:0000255"
FT TOPO_DOM 249..288
FT /note="Extracellular"
FT /evidence="ECO:0000255"
FT TRANSMEM 289..309
FT /note="Helical"
FT /evidence="ECO:0000255"
FT TOPO_DOM 310..326
FT /note="Cytoplasmic"
FT /evidence="ECO:0000255"
FT TRANSMEM 327..347
FT /note="Helical"
FT /evidence="ECO:0000255"
FT TOPO_DOM 348..379
FT /note="Extracellular"
FT /evidence="ECO:0000255"
FT TRANSMEM 380..400
FT /note="Helical"
FT /evidence="ECO:0000255"
FT TOPO_DOM 401..427
FT /note="Cytoplasmic"
FT /evidence="ECO:0000255"
FT TRANSMEM 428..448
FT /note="Helical"
FT /evidence="ECO:0000255"
FT TOPO_DOM 449..459
FT /note="Extracellular"
FT /evidence="ECO:0000255"
FT TRANSMEM 460..480
FT /note="Helical"
FT /evidence="ECO:0000255"
FT TOPO_DOM 481..505
FT /note="Cytoplasmic"
FT /evidence="ECO:0000255"
FT TRANSMEM 506..526
FT /note="Helical"
FT /evidence="ECO:0000255"
FT TOPO_DOM 527..534
FT /note="Extracellular"
FT /evidence="ECO:0000255"
FT TRANSMEM 535..555
FT /note="Helical"
FT /evidence="ECO:0000255"
FT TOPO_DOM 556..596
FT /note="Cytoplasmic"
FT /evidence="ECO:0000255"
SQ SEQUENCE 596 AA; 67262 MW; 8658DCDA5A7D6B64 CRC64;
MTKERMTIDY ENDGDFEYDK NKYKTITTRI KSIEPSEGWL EPSGSVGHIN TIPEAGDVHV
DEHEDRGSSI DDDSRTYLLY FTETRRKLEN RHVQLIAISG VIGTALFVAI GKALYRGGPA
SLLLAFALWC VPILCITVST AEMVCFFPVS SPFLRLATKC VDDSLAVMAS WNFWFLECVQ
IPFEIVSVNT IIHYWRDDYS AGIPLAVQVV LYLLISICAV KYYGEMEFWL ASFKIILALG
LFTFTFITML GGNPEHDRYG FRNYGESPFK KYFPDGNDVG KSSGYFQGFL ACLIQASFTI
AGGEYISMLA GEVKRPRKVL PKAFKQVFVR LTFLFLGSCL CVGIVCSPND PDLTAAINEA
RPGAGSSPYV IAMNNLKIRI LPDIVNIALI TAAFSAGNAY TYCSSRTFYG MALDGYAPKI
FTRCNRHGVP IYSVAISLVW ALVSLLQLNS NSAVVLNWLI NLITASQLIN FVVLCIVYLF
FRRAYHVQQD SLPKLPFRSW GQPYTAIIGL VSCSAMILIQ GYTVFFPKLW NTQDFLFSYL
MVFINIGIYV GYKFIWKRGK DHFKNPHEID FSKELTEIEN HEIESSFEKF QYYSKA
Um primeiro lugar devemos reparar que as linhas começam com um conjunto de duas letras que identifica o tipo de informação que está no resto da linha. Por exemplo, GN é a linha com Gene name, AC é uma linha contendo números de acesso, etc.
O documentação da UniProt contem uma descrição detalhada com estes códigos.
No final de cada bloco de uma proteína encontra-se a sua sequência de amino-ácidos. São as únicas linhas sem código de duas letras (embora a linha introdutória da sequência contenha o código SQ):
...
...
SQ SEQUENCE 440 AA; 49072 MW; DCE9E5C434D51201 CRC64;
MESQQLSQHS PISHGSACAS VTSKEVHTNQ DPLDVSASKT EECEKASTKA NSQQTTTPAS
SAVPENPHHA SPQTAQSHSP QNGPYPQQCM MTQNQANPSG WSFYGHPSMI PYTPYQMSPM
YFPPGPQSQF PQYPSSVGTP LSTPSPESGN TFTDSSSADS DMTSTKKYVR PPPMLTSPND
FPNWVKTYIK FLQNSNLGGI IPTVNGKPVR QITDDELTFL YNTFQIFAPS QFLPTWVKDI
LSVDYTDIMK ILSKSIEKMQ SDTQEANDIV TLANLQYNGS TPADAFETKV TNIIDRLNNN
GIHINNKVAC QLIMRGLSGE YKFLRYTRHR HLNMTVAELF LDIHAIYEEQ QGSRNSKPNY
RRNPSDEKND SRSYTNTTKP KVIARNPQKT NNSKSKTARA HNVSTSNNSP STDNDSISKS
TTEPIQLNNK HDLHLRPETY
Voltando ao exemplo do princípio de cada bloco, as duas primeiras linhas têm a informação que precisamos: o número de acesso e o comprimento da proteína:
ID AGP2_YEAST Reviewed; 596 AA.
AC P38090; D6VQC9;
...
...
Vamos tentar extraír para um dicionário o número de aminoácidos e o número de acesso da primeira proteína da lista obtida anteriormente.
Uma vez que a informação está claramente organizada linha a linha, então podemos para a primeira proteína, começar por separar o bloco de texto por linhas e obter a primeira e a segunda linhas:
# NOTA: continuação do programa anterior!
record = prots[0] # mais tarde aplicaremos a todos os blocos
lines = record.splitlines()
lineID = lines[0]
lineAC = lines[1]
print(lineID)
print(lineAC)
ID AGP2_YEAST Reviewed; 596 AA.
AC P38090; D6VQC9;
Funciona. Vamos agora extraír o que interessa a partir destas linhas.
Se separarmos os elementos da primeira linha pelos espaços o número de aminoácidos está na posição 3!
O número de acesso está na linha AC. O primeiro identificador é o mais atual, os outros são identificadores antigos. A estratégia será aqui separar pelos ; e pelos espaços.
# NOTA: continuação do programa anterior!
record = prots[0] # mais tarde aplicaremos a todos os blocos
lines = record.splitlines()
lineID = lines[0]
lineAC = lines[1]
partsID = lineID.split()
n = partsID[3]
n = int(n) # para forçar que seja um número inteiro
partsAC = lineAC.split(';')
ac = partsAC[0] # isto é AC P38090, é preciso tirar a parte AC
ac = ac.split()
ac = ac[1]
resultado = {'n': n, 'ac':ac}
print(resultado)
{'n': 596, 'ac': 'P38090'}
Resultou. Criámos um dicionário com as duas partes que nos interessavam extraídas.
Desde já um melhoramento: Poddemos aplicar muitas funções de string do Python em cadeia e ainda combinar com a indexação. isto pode-nos poupar o uso de nomes intermédios.
Vejamos uma alternativa:
# NOTA: continuação do programa anterior!
record = prots[0] # mais tarde aplicaremos a todos os blocos
lines = record.splitlines()
lineID = lines[0]
lineAC = lines[1]
ac = lineAC.split(';')[0].split()[1]
n = int(lineID.split()[3])
resultado = {'n': n, 'ac':ac}
print(resultado)
{'n': 596, 'ac': 'P38090'}
Vamos dissecar o processo de obter o ac.
ac = lineAC.split(';')[0].split()[1]
Isto significa:
Dividir a linha
lineACpor;aproveitar o fragmento 0
dividir este por espaços
aproveitar o fragmento 1
Podemos agora criar uma função que possa ser aplicada a qualquer bloco de texto com a informação sobre uma proteína (e não apenas à proteína 0).
Vejamos todo o programa até agora, já testando a nova função com a proteína 0 da lista prots resultantes da primeira função:
data_filename = 'uniprot_scerevisiae.txt'
def read_Uniprot_text(filename):
"""Reads a UniProt text file and splits into a list of protein records."""
with open(filename) as uniprot_file:
whole_file = uniprot_file.read()
records = whole_file.split('//\n')
# remove empty records
records = [p for p in records if len(p) != 0]
# since we know that it is the last one only...
# records.pop(-1)
return records
prots = read_Uniprot_text(data_filename)
def extract_info(record):
lines = record.splitlines()
IDline = lines[0]
ACline = lines[1]
# Extract UniProt AC and number of amino acids
# Example (first two lines):
# ID AB140_YEAST Reviewed; 628 AA.
# AC Q08641; D6W2U2; Q08644;
ac = ACline.split(';',1)[0].split()[1]
n = int(IDline.split()[3])
# Return dictionary of extracted information
return {'ac': ac, 'n': n}
print(f'The number of protein records in "{data_filename}" is {len(prots)}')
print('First protein:')
print(extract_info(prots[0]))
The number of protein records in "uniprot_scerevisiae.txt" is 6049
First protein:
{'ac': 'P38090', 'n': 596}
Podemos agora aplicar a função a todas os blocos, com uma lista em compreensão.
Vamos chamar ao resultado all_prots. Será uma lista de dicionários.
O programa, até este ponto será:
data_filename = 'uniprot_scerevisiae.txt'
def read_Uniprot_text(filename):
"""Reads a UniProt text file and splits into a list of protein records."""
with open(filename) as uniprot_file:
whole_file = uniprot_file.read()
records = whole_file.split('//\n')
# remove empty records
records = [p for p in records if len(p) != 0]
# since we know that it is the last one only...
# records.pop(-1)
return records
prots = read_Uniprot_text(data_filename)
def extract_info(record):
lines = record.splitlines()
IDline = lines[0]
ACline = lines[1]
# Extract UniProt AC and number of amino acids
# Example (first two lines):
# ID AB140_YEAST Reviewed; 628 AA.
# AC Q08641; D6W2U2; Q08644;
ac = ACline.split(';',1)[0].split()[1]
n = int(IDline.split()[3])
# Return dictionary of extracted information
return {'ac': ac, 'n': n}
all_prots = [extract_info(p) for p in prots]
print(f'The number of protein records in "{data_filename}" is {len(prots)}')
# see the first 3, as a test
for p in all_prots[:4]:
print('------------------------------------------')
print(p)
The number of protein records in "uniprot_scerevisiae.txt" is 6049
------------------------------------------
{'ac': 'P38090', 'n': 596}
------------------------------------------
{'ac': 'Q12001', 'n': 544}
------------------------------------------
{'ac': 'P53309', 'n': 568}
------------------------------------------
{'ac': 'P40467', 'n': 964}
8.6. Extração da informação: PTMs#
Vamos agora modificar a função extract_info() com a informação que possa existir no ficheiro da Uniprot sobre PTM.
Aqui está uma pequena parte do meio do ficheiro:
FT MOD_RES 223
FT /note="Phosphoserine"
FT /evidence="ECO:0000244|PubMed:17330950,
FT ECO:0000244|PubMed:18407956, ECO:0000244|PubMed:19779198"
FT MOD_RES 228
FT /note="Phosphoserine"
FT /evidence="ECO:0000244|PubMed:19779198"
FT MOD_RES 232
FT /note="Phosphothreonine"
FT /evidence="ECO:0000244|PubMed:19779198"
FT MOD_RES 244
FT /note="Phosphothreonine"
FT /evidence="ECO:0000244|PubMed:19779198"
FT MOD_RES 570
FT /note="Phosphothreonine; by PKH1 or PKH2"
Podemos consultar a documentação da UniProt sobre as linhas FT.
Parece evidente que a informação sobre resíduos modificados está em linhas começadas por FT MOD_RES e nas linhas seguintes.
Na linha FT MOD_RES está a posição do aminoácido modificado.
Na linha imediatamente a seguir está /note= e o nome da PTM entre ". E ainda, por vezes está o nome da PTM e, a seguir a ; alguma informação adicional.
Para cada proteína (na função extract_info()) queremos
passar por todas as linhas começadas por
FT MOD_RES, extraír daí a posição da PTMna linha seguinte, obter o que está a seguir a
/note=tirar as
"separar por
;aproveitar a primeira parte. Isto será o nome da PTM.
finalmente, criar um dicionário com associação entre a posição e o nome da PTM, para todas as PTMs
e incluír este dicionário no return da função
extract_info()
Como fazer isto por programação?
Temos vários problemas para resolver:
Como passar por todas as linhas respeitantes a uma proteína e procurar as que contêm informação sobre PTM? (fácil,
fore ver se começam porFT MOD_RES)Como extraír a posição? (fácil: separar por espaços e usar o elemento na posição 2)
Como ir para a linha seguinte? (menos fácil, mas
enumerate()permite ter acesso à posição da linha que estamos a tratar, logo também a posição da linha seguinte)Como obter o nome da PTM? (fácil, mais uns
split()s)
Vamos então modificar a função extract_info() e incluír estas ideias:
# ... NOTA: continuação do programa a partir
# da função read_Uniprot_text(filename)
prots = read_Uniprot_text(data_filename)
def extract_info(record):
"""Reads a UniProt text record and returns a dict with extrated information.
The returned dict has the following fields:
'ac': the UniProt Access Id,
'n': the sequence length,
'PTMs': a dictionary that associates the location of PTMs (int, as keys)
with the name of the PTM.
"""
lines = record.splitlines()
IDline = lines[0]
ACline = lines[1]
# Extract UniProt AC and number of amino acids
# Example (first two lines):
# ID AB140_YEAST Reviewed; 628 AA.
# AC Q08641; D6W2U2; Q08644;
ac = ACline.split(';',1)[0].split()[1]
n = int(IDline.split()[3])
# Extract FT lines and process PTM information
#
# Example of phosphorylation lines
# FT MOD_RES 342
# FT /note="Phosphoserine"
# FT /evidence="ECO:0000244|PubMed:18407956,
# FT ECO:0000244|PubMed:19779198"
# NOTE: there are 3 spaces between FT and MOD_RES
PTMs = {}
for i, line in enumerate(lines):
if not line.startswith('FT MOD_RES'):
# skip line if does not start with FT MOD_RES
continue
loc = line.split()[2]
nextline = lines[i+1] # here enumerate() is very usefull
PTMtype = nextline.split('/note=')[1]
PTMtype = PTMtype.strip('"') # strip() takes out the ""
PTMtype = PTMtype.split(';')[0] # the name of the PTM is before the ;
# add to dicionary PTMs
PTMs[loc] = PTMtype
# Return dictionary of extracted information
# including the PTMs dicionary
return {'ac': ac, 'n': n, 'PTMs': PTMs}
all_prots = [extract_info(p) for p in prots]
print(f'The number of protein records in "{data_filename}" is {len(prots)}')
# see the first 4, as a test
for p in all_prots[:5]:
print('------------------------------------------')
print(p)
The number of protein records in "uniprot_scerevisiae.txt" is 6049
------------------------------------------
{'ac': 'P38090', 'n': 596, 'PTMs': {}}
------------------------------------------
{'ac': 'Q12001', 'n': 544, 'PTMs': {}}
------------------------------------------
{'ac': 'P53309', 'n': 568, 'PTMs': {'449': 'Phosphothreonine'}}
------------------------------------------
{'ac': 'P40467', 'n': 964, 'PTMs': {'166': 'Phosphoserine', '186': 'Phosphoserine', '963': 'Phosphoserine'}}
------------------------------------------
{'ac': 'P0CZ17', 'n': 362, 'PTMs': {}}
Aparentemente nem todas as proteínas têm anotações sobre PTMs.
Note-se a utilidade da função enumerate(): podemos ao mesmo tempo passar com o comando for pelas linhas usando o nome line e pelas posições das linhas, usando i (não esquecer que lines é uma lista de linhas).
for i, line in enumerate(lines):
isto foi útil para obter facilmente a “linha seguinte”:
nextline = lines[i+1]
Já se conseguiu muito: agora temos uma lista de 6049 dicionários em que cada um deles tem o número de acesso, o comprimento da proteína e um dicionário com as localizações e o nome de cada PTM da proteína.
O próximo passo é contar as PTM de cada tipo, listar e fazer um gráfico de barras.
8.7. Interlúdio: atribuições múltiplas com *#
Antes de passarmos à fase seguinte, existe um pormenor da atribuição de nomes que pode ser útil. Trata-se da atribuição
múltipla de nomes com a opção de utilizar * para captar o “restante” de uma coleção.
Vejamos um pequeno exemplo de atribuições múltiplas, uma técnica já usada em problemas anteriores:
# atribuição múltipla
m, a = 12, 365
print(m, a)
# atribuição múltipla a partir dos
# elementos de uma lista
s, m, a = [7, 12, 365]
print(s, m, a)
12 365
7 12 365
Aqui mostra-se o facto de uma atribuição poder ser feita à custa dos elementos de uma lista.
Para todos os efeitos, conseguimos assim atribuír nomes a todos os elementos de uma lista:
s, m, a = [7, 12, 365]
# é equivalente a
nums = [7, 12, 365]
s = nums[0]
m = nums[1]
a = nums[2]
Nesta situação, é possível usar, do lado esquerdo do sinal de igual, um *
como prefixo de um dos nomes para captar um conjunto de elementos restantes nas atribuições, na forma de
uma lista mais pequena.
O melhor é ver um exemplo:
s, m, a, *tudo_o_resto = [7, 12, 365, 1, 3, 5, 7, 9, 11]
print(s)
print(m)
print(a)
print(tudo_o_resto)
7
12
365
[1, 3, 5, 7, 9, 11]
Neste exemplo, pelo facto de termos usado um * antes do nome tudo_o_resto
fez com este nome fosse atribuído à parte restante da lista que serviu de atribuição.
Dica
Esta atribuição múltipla com * é muito útil quando não sabemos
exatamente qual o comprimento da lista que fornece os valores e estamos
interessados apenas em alguns dos primeiros valores.
O nome que é usado com * como prefixo é sempre atribuído a uma lista.
Esta pode ter apenas um elemento ou até uma lista vazia.
Mas este mecanismo pode até ser usado sem ser no nome final:
s, m, a, *o_meio, ultimo = [7, 12, 365, 1, 3, 5, 7, 9, 11]
print(s)
print(m)
print(a)
print(o_meio)
print(ultimo)
7
12
365
[1, 3, 5, 7, 9]
11
Um outro exemplo:
*lixo, antepenúltimo, penúltimo, último = [7, 12, 365, 1, 3, 5, 7, 9, 11]
print(antepenúltimo)
print(penúltimo)
print(último)
7
9
11
E ainda outro…
primeiro, *lixo, antepenúltimo, penúltimo, último = [7, 12, 365, 1, 3, 5, 7, 9, 11]
print(primeiro)
print(antepenúltimo)
print(penúltimo)
print(último)
print(lixo)
7
7
9
11
[12, 365, 1, 3, 5]
Este mecanismo pode-nos ajudar a simplificar um pouco alguns passos da função
extract_info():
Em vez de
lines = record.splitlines()
IDline = lines[0]
ACline = lines[1]
Podemos escrever apenas
IDline, ACline, *otherlines = record.splitlines()
A função .splitlines() gera uma lista, logo podemos imediatamente usar
uma atribuição múltipla para atribuír nomes a várias linhas que estejamos interessados.
Note-se que otherlines seria um nome de uma lista contendo todas as linhas
menos as duas primeiras. O que significa que, mais à frente, quando procuramos
linhas começadas por FT, devemos fazer
for line in otherlines:
Problema
Como poderíamos substituír a obtenção de n nesta parte do program
n = int(IDline.split()[3])
por uma atribuição múltipla para isolar o n no meio dos outros
elementos na mesma linha?
Repare que a linha ID tem o seguinte aspeto:
ID AB140_YEAST Reviewed; 628 AA.
Solução
*useless, n, final = IDline.split()
n = int(n)
Não sendo mais compacto, é mais simples de entender
8.8. Contagem dos tipos de PTM#
Vamos agora continuar o programa de forma contar os vários tipos de PTM que existem, já
“extraídos” na lista de dicionários all_prots.
Atenção
Nos passos seguintes vamos ampliar o programa que já existe, até
ao cálculo da lista all_prots. Esta lista já tem de existir para o resto
funcionar.
Recorde-se que a lista all_prots contem dicionários.
Cada dicionário contem
uma chave PTMs, a qual tem como valor um outro dicionário que associa a posição
de cada PTM ao seu nome.
{'ac': 'P40467', 'n': 964, 'PTMs': {'166': 'Phosphoserine', '186': 'Phosphoserine', '963': 'Phosphoserine'}}
Como contar as PTM em todas as proteínas pelo seu nome?
Podemos criar, mais uma vez, um novo dicionário que associa o nome de cada PTM à sua contagem. Vamos chamar
PTM_counts.
A ideia será passar por toda a lista all_prots (com for p in all_prots),
obter os nomes das PTM de cada proteína (com p['PTMs'].values()) e depois
ir adicionando +1 à contagem existente no dicionário de contagens.
Se ainda não existir o nome de uma PTM no dicionário, na primeira vez que é colocado deve ter o valor 1, a primeira contagem.
Vejamos como fica:
PTM_counts = {}
for p in all_prots:
PTMs = p['PTMs']
# just skip if no PTMs
if len(PTMs) == 0:
continue
for ptmtype in PTMs.values():
if ptmtype in PTM_counts:
PTM_counts[ptmtype] = PTM_counts[ptmtype] + 1
else:
PTM_counts[ptmtype] = 1
print(PTM_counts)
{'Phosphothreonine': 1027, 'Phosphoserine': 5170, 'Phosphotyrosine': 55, 'N-acetylserine': 325, 'N-formylmethionine': 1, 'N6-(pyridoxal phosphate)lysine': 46, 'Pros-8alpha-FAD histidine': 1, 'N-acetylmethionine': 106, 'N6-acetyllysine': 32, 'N-acetylthreonine': 12, "O-(pantetheine 4'-phosphoryl)serine": 3, 'N6-carboxylysine': 4, 'Cysteine methyl ester': 23, 'Asymmetric dimethylarginine': 26, 'Phosphohistidine': 4, 'Hypusine': 2, 'N-acetylalanine': 37, 'N6-butyryllysine': 5, 'N6-methyllysine': 22, 'N6-succinyllysine': 11, 'N6,N6-dimethyllysine': 6, 'Tele-8alpha-FAD histidine': 2, '2,3-didehydroalanine (Cys)': 1, '4-aspartylphosphate': 3, 'Lysine derivative': 3, 'N5-methylglutamine': 4, 'N6,N6,N6-trimethyllysine': 17, 'Omega-N-methylarginine': 15, 'N,N-dimethylproline': 4, 'N6-glutaryllysine': 1, 'N6-malonyllysine': 2, 'Diphthamide': 1, 'N6-biotinyllysine': 5, 'N5-methylarginine': 2, 'N6-lipoyllysine': 4, 'Pyruvic acid (Ser)': 3, 'Pros-methylhistidine': 1, 'S-glutathionyl cysteine': 3, 'Cysteine sulfinic acid (-SO2H)': 1, 'N6-propionyllysine': 2, 'S-(dipyrrolylmethanemethyl)cysteine': 1, 'N,N,N-trimethylglycine': 1, 'Lysine methyl ester': 1, 'Dimethylated arginine': 1, 'Leucine methyl ester': 2, '3,4-dihydroxyproline': 2, 'N-acetylvaline': 2, 'Thiazolidine linkage to a ring-opened DNA abasic': 1, '1-thioglycine': 1, 'S-methylcysteine': 1}
Funcionou, embora não seja a maneira mais elegante de apresentar as contagens.
Na realidade o ideal seria ordenar por ordem decrescente, de forma a apresentar na forma de uma tabela em que saberíamos facilemnte quais as PTM mais abundantes e menos abundantes.
O problema é que os dicionários não têm uma ordem dos seus elementos implícita.
As listas têm e é fácil transformar um dicionário numa lista de pares (chave:valor):
PTM_counts = {}
for p in all_prots:
PTMs = p['PTMs']
# just skip if no PTMs
if len(PTMs) == 0:
continue
for ptmtype in PTMs.values():
if ptmtype in PTM_counts:
PTM_counts[ptmtype] = PTM_counts[ptmtype] + 1
else:
PTM_counts[ptmtype] = 1
# print(PTM_counts)
list_PTM_counts = list(PTM_counts.items())
print(list_PTM_counts)
[('Phosphothreonine', 1027), ('Phosphoserine', 5170), ('Phosphotyrosine', 55), ('N-acetylserine', 325), ('N-formylmethionine', 1), ('N6-(pyridoxal phosphate)lysine', 46), ('Pros-8alpha-FAD histidine', 1), ('N-acetylmethionine', 106), ('N6-acetyllysine', 32), ('N-acetylthreonine', 12), ("O-(pantetheine 4'-phosphoryl)serine", 3), ('N6-carboxylysine', 4), ('Cysteine methyl ester', 23), ('Asymmetric dimethylarginine', 26), ('Phosphohistidine', 4), ('Hypusine', 2), ('N-acetylalanine', 37), ('N6-butyryllysine', 5), ('N6-methyllysine', 22), ('N6-succinyllysine', 11), ('N6,N6-dimethyllysine', 6), ('Tele-8alpha-FAD histidine', 2), ('2,3-didehydroalanine (Cys)', 1), ('4-aspartylphosphate', 3), ('Lysine derivative', 3), ('N5-methylglutamine', 4), ('N6,N6,N6-trimethyllysine', 17), ('Omega-N-methylarginine', 15), ('N,N-dimethylproline', 4), ('N6-glutaryllysine', 1), ('N6-malonyllysine', 2), ('Diphthamide', 1), ('N6-biotinyllysine', 5), ('N5-methylarginine', 2), ('N6-lipoyllysine', 4), ('Pyruvic acid (Ser)', 3), ('Pros-methylhistidine', 1), ('S-glutathionyl cysteine', 3), ('Cysteine sulfinic acid (-SO2H)', 1), ('N6-propionyllysine', 2), ('S-(dipyrrolylmethanemethyl)cysteine', 1), ('N,N,N-trimethylglycine', 1), ('Lysine methyl ester', 1), ('Dimethylated arginine', 1), ('Leucine methyl ester', 2), ('3,4-dihydroxyproline', 2), ('N-acetylvaline', 2), ('Thiazolidine linkage to a ring-opened DNA abasic', 1), ('1-thioglycine', 1), ('S-methylcysteine', 1)]
Agora temos uma lista, mas não está ordenada por contagens.
Como ordenar esta lista?
Repare-se que os elementos da lista são pares de objetos e queremos ordenar a lista pelo segundo elemento do par de objetos.
Existe uma função para gerar uma sequência de valores ordenados a partir de uma lista: a função
sorted()
Mas esta função pode receber como argumento key uma outra função que indique
qual exatamente o valor que deve ser ordenado, calculado a partir de cada elemento.
Isto é útil, neste caso. Precisamos de uma função que “aponte” para o segundo elemento
de cada par para que a função sorted() saiba o que fazer.
Mas temos de escrever essa função.
É uma função muito simples que recebe um par de valores e devolve o segundo elemento do par.
# sort function returning the second element of a pair of values
def second(pair):
return pair[1]
Vejamos como aplicar esta função:
# sort function returning the second element of a pair of values
def second(pair):
return pair[1]
ordered_PTM_counts = sorted(list_PTM_counts, key=second, reverse=True)
print(ordered_PTM_counts)
[('Phosphoserine', 5170), ('Phosphothreonine', 1027), ('N-acetylserine', 325), ('N-acetylmethionine', 106), ('Phosphotyrosine', 55), ('N6-(pyridoxal phosphate)lysine', 46), ('N-acetylalanine', 37), ('N6-acetyllysine', 32), ('Asymmetric dimethylarginine', 26), ('Cysteine methyl ester', 23), ('N6-methyllysine', 22), ('N6,N6,N6-trimethyllysine', 17), ('Omega-N-methylarginine', 15), ('N-acetylthreonine', 12), ('N6-succinyllysine', 11), ('N6,N6-dimethyllysine', 6), ('N6-butyryllysine', 5), ('N6-biotinyllysine', 5), ('N6-carboxylysine', 4), ('Phosphohistidine', 4), ('N5-methylglutamine', 4), ('N,N-dimethylproline', 4), ('N6-lipoyllysine', 4), ("O-(pantetheine 4'-phosphoryl)serine", 3), ('4-aspartylphosphate', 3), ('Lysine derivative', 3), ('Pyruvic acid (Ser)', 3), ('S-glutathionyl cysteine', 3), ('Hypusine', 2), ('Tele-8alpha-FAD histidine', 2), ('N6-malonyllysine', 2), ('N5-methylarginine', 2), ('N6-propionyllysine', 2), ('Leucine methyl ester', 2), ('3,4-dihydroxyproline', 2), ('N-acetylvaline', 2), ('N-formylmethionine', 1), ('Pros-8alpha-FAD histidine', 1), ('2,3-didehydroalanine (Cys)', 1), ('N6-glutaryllysine', 1), ('Diphthamide', 1), ('Pros-methylhistidine', 1), ('Cysteine sulfinic acid (-SO2H)', 1), ('S-(dipyrrolylmethanemethyl)cysteine', 1), ('N,N,N-trimethylglycine', 1), ('Lysine methyl ester', 1), ('Dimethylated arginine', 1), ('Thiazolidine linkage to a ring-opened DNA abasic', 1), ('1-thioglycine', 1), ('S-methylcysteine', 1)]
Agora sim, temos uma lista ordenada.
Mas é muito mais elegante apresentar em linhas separadas:
# sort function returning the second element of a pair of values
def second(pair):
return pair[1]
ordered_PTM_counts = sorted(list_PTM_counts, key=second, reverse=True)
# let's look at the results
for PTMtype, count in ordered_PTM_counts:
print(PTMtype, count)
Phosphoserine 5170
Phosphothreonine 1027
N-acetylserine 325
N-acetylmethionine 106
Phosphotyrosine 55
N6-(pyridoxal phosphate)lysine 46
N-acetylalanine 37
N6-acetyllysine 32
Asymmetric dimethylarginine 26
Cysteine methyl ester 23
N6-methyllysine 22
N6,N6,N6-trimethyllysine 17
Omega-N-methylarginine 15
N-acetylthreonine 12
N6-succinyllysine 11
N6,N6-dimethyllysine 6
N6-butyryllysine 5
N6-biotinyllysine 5
N6-carboxylysine 4
Phosphohistidine 4
N5-methylglutamine 4
N,N-dimethylproline 4
N6-lipoyllysine 4
O-(pantetheine 4'-phosphoryl)serine 3
4-aspartylphosphate 3
Lysine derivative 3
Pyruvic acid (Ser) 3
S-glutathionyl cysteine 3
Hypusine 2
Tele-8alpha-FAD histidine 2
N6-malonyllysine 2
N5-methylarginine 2
N6-propionyllysine 2
Leucine methyl ester 2
3,4-dihydroxyproline 2
N-acetylvaline 2
N-formylmethionine 1
Pros-8alpha-FAD histidine 1
2,3-didehydroalanine (Cys) 1
N6-glutaryllysine 1
Diphthamide 1
Pros-methylhistidine 1
Cysteine sulfinic acid (-SO2H) 1
S-(dipyrrolylmethanemethyl)cysteine 1
N,N,N-trimethylglycine 1
Lysine methyl ester 1
Dimethylated arginine 1
Thiazolidine linkage to a ring-opened DNA abasic 1
1-thioglycine 1
S-methylcysteine 1
Agora sim, vemos que as fosforilações são as PTM mais prevalentes, pelo menos levando em conta as anotações na UniProtKB relativamente às proteínas de levedura S. cerevisiae.
Conseguimos extraír a informação em que estávamos interessados a partir de um ficheiro de texto que segue um formato rígido particular a um portal de Bioinformática.
Note-se, no desenvolvimento deste programa, o papel essencial das coleções da linguagem Python, listas e dicionários, para armazenar a informação obtida e também o papel das funções associadas a strings
O programa todo até este ponto é o seguinte:
data_filename = 'uniprot_scerevisiae.txt'
def read_Uniprot_text(filename):
"""Reads a UniProt text file and splits into a list of protein records."""
with open(filename) as uniprot_file:
whole_file = uniprot_file.read()
records = whole_file.split('//\n')
# remove empty records
records = [p for p in records if len(p) != 0]
# since we know that it is the last one only...
# records.pop(-1)
return records
prots = read_Uniprot_text(data_filename)
def extract_info(record):
"""Reads a UniProt text record and returns a dict with extrated information.
The returned dict has the following fields:
'ac': the UniProt Access Id,
'n': the sequence length,
'PTMs': a dictionary that associates the location of PTMs (int, as keys)
with the name of the PTM.
"""
IDline, ACline, *otherlines = record.splitlines()
# Extract UniProt AC and number of amino acids
# Example (first two lines):
# ID AB140_YEAST Reviewed; 628 AA.
# AC Q08641; D6W2U2; Q08644;
ac = ACline.split(';',1)[0].split()[1]
n = int(IDline.split()[3])
# Extract FT lines and process PTM information
#
# Example of phosphorylation lines
# FT MOD_RES 342
# FT /note="Phosphoserine"
# FT /evidence="ECO:0000244|PubMed:18407956,
# FT ECO:0000244|PubMed:19779198"
PTMs = {}
for i, line in enumerate(otherlines):
if line.startswith('FT MOD_RES'):
FTcode, MOD_RES, loc, *rest = line.split()
nextline = otherlines[i+1]
PTMtype = nextline.split('/note=')[1]
PTMtype = PTMtype.strip('"')
PTMtype = PTMtype.split(';')[0]
PTMs[loc] = PTMtype
# Return dictionary of extracted information
return {'ac': ac, 'n': n, 'PTMs': PTMs}
all_prots = [extract_info(p) for p in prots]
PTM_counts = {}
for p in all_prots:
PTMs = p['PTMs']
# just skip if no PTMs
if len(PTMs) == 0:
continue
for ptmtype in PTMs.values():
if ptmtype in PTM_counts:
PTM_counts[ptmtype] = PTM_counts[ptmtype] + 1
else:
PTM_counts[ptmtype] = 1
list_PTM_counts = list(PTM_counts.items())
# sort function returning the second element of a pair of values
def second(pair):
return pair[1]
ordered_PTM_counts = sorted(list_PTM_counts, key=second, reverse=True)
# Output of PTM information
print(f'The number of protein records in "{data_filename}" is {len(prots)}')
for PTMtype, count in ordered_PTM_counts:
print(PTMtype, count)
The number of protein records in "uniprot_scerevisiae.txt" is 6049
Phosphoserine 5170
Phosphothreonine 1027
N-acetylserine 325
N-acetylmethionine 106
Phosphotyrosine 55
N6-(pyridoxal phosphate)lysine 46
N-acetylalanine 37
N6-acetyllysine 32
Asymmetric dimethylarginine 26
Cysteine methyl ester 23
N6-methyllysine 22
N6,N6,N6-trimethyllysine 17
Omega-N-methylarginine 15
N-acetylthreonine 12
N6-succinyllysine 11
N6,N6-dimethyllysine 6
N6-butyryllysine 5
N6-biotinyllysine 5
N6-carboxylysine 4
Phosphohistidine 4
N5-methylglutamine 4
N,N-dimethylproline 4
N6-lipoyllysine 4
O-(pantetheine 4'-phosphoryl)serine 3
4-aspartylphosphate 3
Lysine derivative 3
Pyruvic acid (Ser) 3
S-glutathionyl cysteine 3
Hypusine 2
Tele-8alpha-FAD histidine 2
N6-malonyllysine 2
N5-methylarginine 2
N6-propionyllysine 2
Leucine methyl ester 2
3,4-dihydroxyproline 2
N-acetylvaline 2
N-formylmethionine 1
Pros-8alpha-FAD histidine 1
2,3-didehydroalanine (Cys) 1
N6-glutaryllysine 1
Diphthamide 1
Pros-methylhistidine 1
Cysteine sulfinic acid (-SO2H) 1
S-(dipyrrolylmethanemethyl)cysteine 1
N,N,N-trimethylglycine 1
Lysine methyl ester 1
Dimethylated arginine 1
Thiazolidine linkage to a ring-opened DNA abasic 1
1-thioglycine 1
S-methylcysteine 1
8.9. Gráficos#
A tabela que conseguimos obter na secção anterior já é muito interessante.
Ficámos a saber que a fosforilação de serinas tem 6049 anotações, enquanto que a PTM exótica S-metilcisteína tem uma única anotação (ela e outras 13).
Mas um gráfico causaria muito mais impressão. Vamos fazer um gráfico de barras com as contagens de PTM.
Para a linguagem Python existem vários módulos destinados a obter gráficos num
program. Um dos mais conhecidos é o matplotlib, uma da “bibliotecas” gráficas
mais versáteis e populares (e também um pouco intimidante) da linguagem Python.
O site oficial da matplotlib dá uma ideia da versatilidade da biblioteca, tendo uma galeria extensa de exemplos.
Uma outra biblioteca digna de nota é o projeto seaborn que é
baseado na matplotlib e inclui “melhoramentos” estéticos e novos tipos de gráficos, especialemnte da área da estatística.
Ampliando o programa, vamos combinar os dois módulos, criando um gráfico de barras com as contagens da PTM, embora restringindo àquelas que têm pelo menos 10 contagens:
from matplotlib import pyplot as plt
import seaborn as sns
sns.set(style="darkgrid")
f, ax = plt.subplots(figsize=(6,9))
types = [t for t,c in ordered_PTM_counts if c > 10]
counts = [c for t,c in ordered_PTM_counts if c > 10]
bp = sns.barplot(y=types, x=counts, orient='h', log=True, palette='tab10')
plt.show()
Embora seja uma pequena ampliação do programa, é preciso explicar um pouco a utilização destas duas bibliotecas, de uma forma muitíssimo sumária, uma vez que a sua funcionalidade é muito vasta.
Após os dois imports necessários para se utilizar as bibliotecas, a função
seaborn.set() introduz um estilo de gráficos particular, o “darkgrid” (existem vários).
É uma questão estética (e de gosto pessoal).
A função subplots() é uma função importante que prepara uma figura se tiver vários painéis, o que não é
o caso e, de entre outras opções, permite controlar o tamanho total da figura, com figsize (unidades em polegadas).
Depois disto é apenas necessário preparar duas listas, uma com os nomes das barras e
outra com as alturas da barras e usar a função seaborn.barplot() usando estas duas
listas como argumentos.
As listas foram criadas como listas em compreensão a partir da lista ordered_PTM_counts,
filtrando as PTM com mais de 10 contagens.
Mais 3 pormenores finais na função barplot():
orient='h'desenha barras horizontais e não (as mais habituais) barras verticaispalette='tab10'escolhe um mapa de cores a usar nas barras. Há inúmeras possibilidades. Ver por exemplo esta páginafinalmente, last but not least,
log=True, faz a escala de contagens ser logarítmica. Isto é necessário, caso contrário as fosforilações dominariam completamente o gráfico e não conseguiríamos ver as PTM menos abundantes que teríam barras perto de 0.
8.10. Programa completo#
data_filename = 'uniprot_scerevisiae.txt'
def read_Uniprot_text(filename):
"""Reads a UniProt text file and splits into a list of protein records."""
with open(filename) as uniprot_file:
whole_file = uniprot_file.read()
records = whole_file.split('//\n')
# remove empty records
records = [p for p in records if len(p) != 0]
# since we know that it is the last one only...
# records.pop(-1)
return records
prots = read_Uniprot_text(data_filename)
def extract_info(record):
"""Reads a UniProt text record and returns a dict with extrated information.
The returned dict has the following fields:
'ac': the UniProt Access Id,
'n': the sequence length,
'PTMs': a dictionary that associates the location of PTMs (int, as keys)
with the name of the PTM.
"""
IDline, ACline, *otherlines = record.splitlines()
# Extract UniProt AC and number of amino acids
# Example (first two lines):
# ID AB140_YEAST Reviewed; 628 AA.
# AC Q08641; D6W2U2; Q08644;
ac = ACline.split(';',1)[0].split()[1]
n = int(IDline.split()[3])
# Extract FT lines and process PTM information
#
# Example of phosphorylation lines
# FT MOD_RES 342
# FT /note="Phosphoserine"
# FT /evidence="ECO:0000244|PubMed:18407956,
# FT ECO:0000244|PubMed:19779198"
PTMs = {}
for i, line in enumerate(otherlines):
if line.startswith('FT MOD_RES'):
FTcode, MOD_RES, loc, *rest = line.split()
nextline = otherlines[i+1]
PTMtype = nextline.split('/note=')[1]
PTMtype = PTMtype.strip('"')
PTMtype = PTMtype.split(';')[0]
PTMs[loc] = PTMtype
# Return dictionary of extracted information
return {'ac': ac, 'n': n, 'PTMs': PTMs}
all_prots = [extract_info(p) for p in prots]
PTM_counts = {}
for p in all_prots:
PTMs = p['PTMs']
# just skip if no PTMs
if len(PTMs) == 0:
continue
for ptmtype in PTMs.values():
if ptmtype in PTM_counts:
PTM_counts[ptmtype] = PTM_counts[ptmtype] + 1
else:
PTM_counts[ptmtype] = 1
list_PTM_counts = list(PTM_counts.items())
# sort function returning the second element of a pair of values
def second(pair):
return pair[1]
ordered_PTM_counts = sorted(list_PTM_counts, key=second, reverse=True)
# Output of PTM information
print(f'The number of protein records in "{data_filename}" is {len(prots)}')
for PTMtype, count in ordered_PTM_counts:
print(PTMtype, count)
# Bar plot of PTM counts
from matplotlib import pyplot as plt
import seaborn as sns
sns.set(style="darkgrid")
f, ax = plt.subplots(figsize=(6,9))
types = [t for t,c in ordered_PTM_counts if c > 10]
counts = [c for t,c in ordered_PTM_counts if c > 10]
bp = sns.barplot(y=types, x=counts, orient='h', log=True, palette='tab10')
plt.show()
The number of protein records in "uniprot_scerevisiae.txt" is 6049
Phosphoserine 5170
Phosphothreonine 1027
N-acetylserine 325
N-acetylmethionine 106
Phosphotyrosine 55
N6-(pyridoxal phosphate)lysine 46
N-acetylalanine 37
N6-acetyllysine 32
Asymmetric dimethylarginine 26
Cysteine methyl ester 23
N6-methyllysine 22
N6,N6,N6-trimethyllysine 17
Omega-N-methylarginine 15
N-acetylthreonine 12
N6-succinyllysine 11
N6,N6-dimethyllysine 6
N6-butyryllysine 5
N6-biotinyllysine 5
N6-carboxylysine 4
Phosphohistidine 4
N5-methylglutamine 4
N,N-dimethylproline 4
N6-lipoyllysine 4
O-(pantetheine 4'-phosphoryl)serine 3
4-aspartylphosphate 3
Lysine derivative 3
Pyruvic acid (Ser) 3
S-glutathionyl cysteine 3
Hypusine 2
Tele-8alpha-FAD histidine 2
N6-malonyllysine 2
N5-methylarginine 2
N6-propionyllysine 2
Leucine methyl ester 2
3,4-dihydroxyproline 2
N-acetylvaline 2
N-formylmethionine 1
Pros-8alpha-FAD histidine 1
2,3-didehydroalanine (Cys) 1
N6-glutaryllysine 1
Diphthamide 1
Pros-methylhistidine 1
Cysteine sulfinic acid (-SO2H) 1
S-(dipyrrolylmethanemethyl)cysteine 1
N,N,N-trimethylglycine 1
Lysine methyl ester 1
Dimethylated arginine 1
Thiazolidine linkage to a ring-opened DNA abasic 1
1-thioglycine 1
S-methylcysteine 1